


ร่างกายผิดปกติ เจ็บไข้ได้ป่วยแบบหาสาเหตุไม่ได้ ท้องโต ตับโต ม้ามโต เลือดออกง่าย กระดูกหักง่าย ฯลฯ ทุกอาการที่ว่ามาถ้าทำความเข้าใจไม่มากพออาจจะตีความไปว่าเป็นโรคกรรมเก่าซะทีเดียว แต่จริง ๆ แล้วอาจมาจากโรคทาง “พันธุกรรม” ที่ชื่อว่า “โรคโกเช่ร์” หนึ่งในโรคหายากที่หลายคนอาจจะไม่คุ้นชื่อ แต่สามารถรักษาให้อาการดีขึ้น เพื่อคุณภาพชีวิตที่ดีขึ้นได้
รู้หรือไม่ว่า“โรคหายาก” ถือเป็นกลุ่มโรคที่พบได้น้อยแต่มีอยู่จริง ในประเทศไทยมีผู้ป่วยกว่า 3.5 ล้านคน! จากโรคหายากกว่า 7,000 โรคที่มีอยู่บนโลกใบนี้ หนึ่งในนั้น คือ โรคโกเช่ร์ (Gaucher Disease) ซึ่งมีอุบัติการณ์ผู้ป่วยราว 1 คนต่อ 100,000 คนเท่านั้น หลายคนจึงไม่รู้จักและไม่เคยได้ยินชื่อโรคนี้มาก่อน จริง ๆ แล้ว National Gaucher Foundation เคยจัดทำหนังสั้นแนวเศร้าปนตลกขนขัน เรื่อง “One of Those Dates” ที่หยิบยกเรื่องราวของโรคโกเช่ร์ออกมาเป็นส่วนหนึ่งของบทสนทนาของตัวละครในเดทแรก แต่ “โรคโกเช่ร์” ก็ยังไม่เป็นที่รู้จักในวงกว้าง ทำให้ผู้ป่วยส่วนใหญ่ใช้ชีวิตอยู่กับโรค ที่สังคมไม่รู้จักและไม่มีความเข้าใจ เพราะอาการดูคล้าย ๆ กับโรคทั่วไป จากรายงานต่างประเทศพบว่าผู้ป่วยโรคโกเช่ร์จะต้องเปลี่ยนแพทย์ไปเรื่อย ๆ โดยเฉลี่ยถึง 7 คน และใช้ระยะเวลานานเป็น 10 ปี กว่าจะวินิจฉัยโรคพบ แถมค่าใช้จ่ายในการรักษาก็สูงลิบ อยู่ที่ราว 2 ล้านบาทต่อปี
เสียงจากคุณหมอซึ่งทำงานคลุกคลีกับผู้ป่วยมายาวนาน ศ.พญ. ดวงฤดี วัฒนศิริชัยกุล หัวหน้าสาขาเวชพันธุศาสตร์ ภาควิชากุมารเวชศาสตร์ โรงพยาบาลรามาธิบดี มหาวิทยาลัยมหิดล กล่าวว่า “โรคนี้ซ่อนตัวในหน่วยพันธุกรรมตั้งแต่กำเนิด มักเกิดกับผู้ที่มีพ่อและแม่ที่เป็นพาหะทั้งคู่ โดยไม่ทราบว่าตนเป็นพาหะ เนื่องจากพาหะไม่แสดงอาการ แต่สามารถถ่ายทอดยีนผิดปกติไปยังลูกได้ ทำให้มีโอกาสมีลูกเป็นโรคได้ร้อยละ 25 ของทุกการตั้งครรภ์ โดยอาการของโรค โกเช่ร์ สามารถสังเกตได้จากรูปร่างที่เริ่มผิดปกติ และแสดงอาการเจ็บป่วย เช่น ท้องโตเพราะตับ ม้ามโต มีอาการทางระบบเลือด เกิดภาวะซีด เกล็ดเลือดต่ำ ทำให้เป็นแผลฟกช้ำและเลือดออกง่ายกว่าปกติ อาการปวดกระดูก มีภาวะกระดูกบาง กระดูกหักง่าย ผู้ป่วยเด็กบางรายอาจมีปัญหาเรื่องการเจริญเติบโตช้า มีความผิดปกติทางพัฒนาการด้านสติปัญญา มีอาการลมชัก เป็นต้น ปัจจุบันการรักษาโรคโกเช่ร์ในประเทศไทย มีอยู่ 2 วิธีซึ่งมีประสิทธิภาพ คือการใช้ยาเอนไซม์ทดแทนเป็นหลัก และการใช้ยาเอนไซม์ทดแทนแล้วปลูกถ่ายไขกระดูกร่วมด้วย”

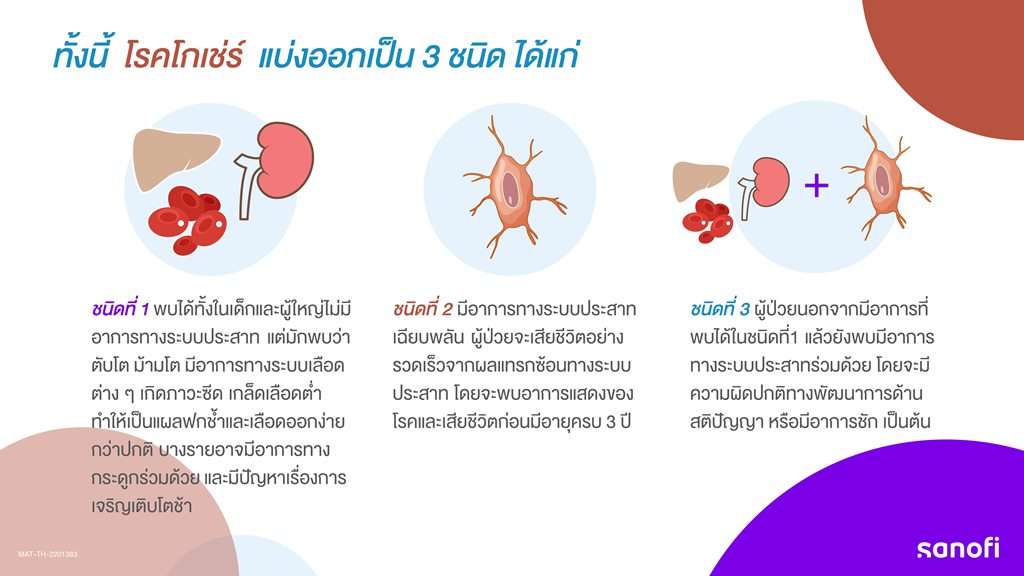
ทั้งนี้ โรคโกเช่ร์ แบ่งออกเป็น 3 ชนิด ได้แก่
ชนิดที่ 1 พบได้ทั้งในเด็กและผู้ใหญ่ไม่มีอาการทางระบบประสาท แต่มักพบว่า ตับโต ม้ามโต มีอาการทางระบบเลือดต่าง ๆ เกิดภาวะซีด เกล็ดเลือดต่ำ ทำให้เป็นแผลฟกช้ำและเลือดออกง่ายกว่าปกติ บางรายอาจมีอาการทางกระดูกร่วมด้วย และมีปัญหาเรื่องการเจริญเติบโตช้า
ชนิดที่ 2 มีอาการทางระบบประสาทเฉียบพลัน ผู้ป่วยจะเสียชีวิตอย่างรวดเร็วจากผลแทรกซ้อนทางระบบประสาท โดยจะพบอาการแสดงของโรคและเสียชีวิตก่อนมีอายุครบ 3 ปี
ชนิดที่ 3 ผู้ป่วยนอกจากมีอาการที่พบได้ในชนิดที่1 แล้วยังพบมีอาการทางระบบประสาทร่วมด้วย โดยจะมีความผิดปกติทางพัฒนาการด้านสติปัญญา หรือมีอาการชัก เป็นต้น
สิ่งสำคัญในการส่งต่อความช่วยเหลือให้แก่ผู้ป่วยคือ การสร้างการรับรู้ถึงการมีอยู่ของโรค การวินิจฉัยและการเข้าถึงการรักษาอย่างรวดเร็ว ตลอดระยะเวลากว่า 20 ปีที่ซาโนฟี่ ประเทศไทย ในฐานะบริษัทชั้นนำด้านสุขภาพระดับโลก พร้อมด้วยสมาคมพันธุศาสตร์แห่งประเทศไทย สมาคมเวชพันธุศาสตร์และจีโนมิกส์ทางการแพทย์ (สวพจ.) สมาคมโลหิตวิทยาแห่งประเทศไทย มูลนิธิเพื่อผู้ป่วยโรคหายาก มูลนิธิโรคพันธุกรรมแอลเอสดี มุ่งมั่นที่จะร่วมเป็นส่วนหนึ่งในการสร้างความเปลี่ยนแปลงให้แก่โรคหายากและยืนหยัดและอยู่เคียงข้างกับกลุ่มผู้ป่วยโรคโกเช่ร์ สร้างการตระหนักรู้ และขับเคลื่อนให้เกิดการเข้าถึงการรักษาโรคโกเช่ร์ ส่งผลให้โรคโกเช่ร์เป็นโรคพันธุกรรมเมแทบอลิกกลุ่มแรก ที่สามารถเข้าสู่ระบบหลักประกันสุขภาพแห่งชาติ ในระบบสาธารณสุขไทย และทำให้ผู้ป่วยหลายชีวิตมีคุณภาพชีวิตที่ดีขึ้น สามารถใช้ชีวิตได้อย่างปกติสุข โดยจะไม่มีใครถูกทิ้งไว้ข้างหลังอีกต่อไป...



.jpg)
.jpg)









